The MHRA was pleased to participate in a workshop on 29 June 2026, organised by the British Embassy in Berne, which brought together senior leaders from the biopharma and space sectors, academia, regulatory bodies and funding organisations across the UK and Switzerland.
The discussions highlighted the growing opportunities for collaboration between industry and academia to advance space-enabled life sciences. Importantly, the conversation extended beyond the manufacturing of medicines in orbit, exploring the wider ecosystem and how microgravity research could unlock new scientific insights, accelerate innovation and support the development of future healthcare technologies.
Meeting purpose
1. Identify opportunities: several promising use cases in cell behaviour, tissue models, and protein science are progressing rapidly and are among the areas closest to delivering real-world impact.
2. Enable adoption: continued generation of proof-of-concept data will help build confidence, while greater regulatory clarity, skills development, and awareness can further accelerate adoption.
3. Targeted solutions: highlight the value of targeted pilot projects, knowledge-exchange programmes, and collaborative consortium-style partnerships to help translate innovation into practice
Key insights
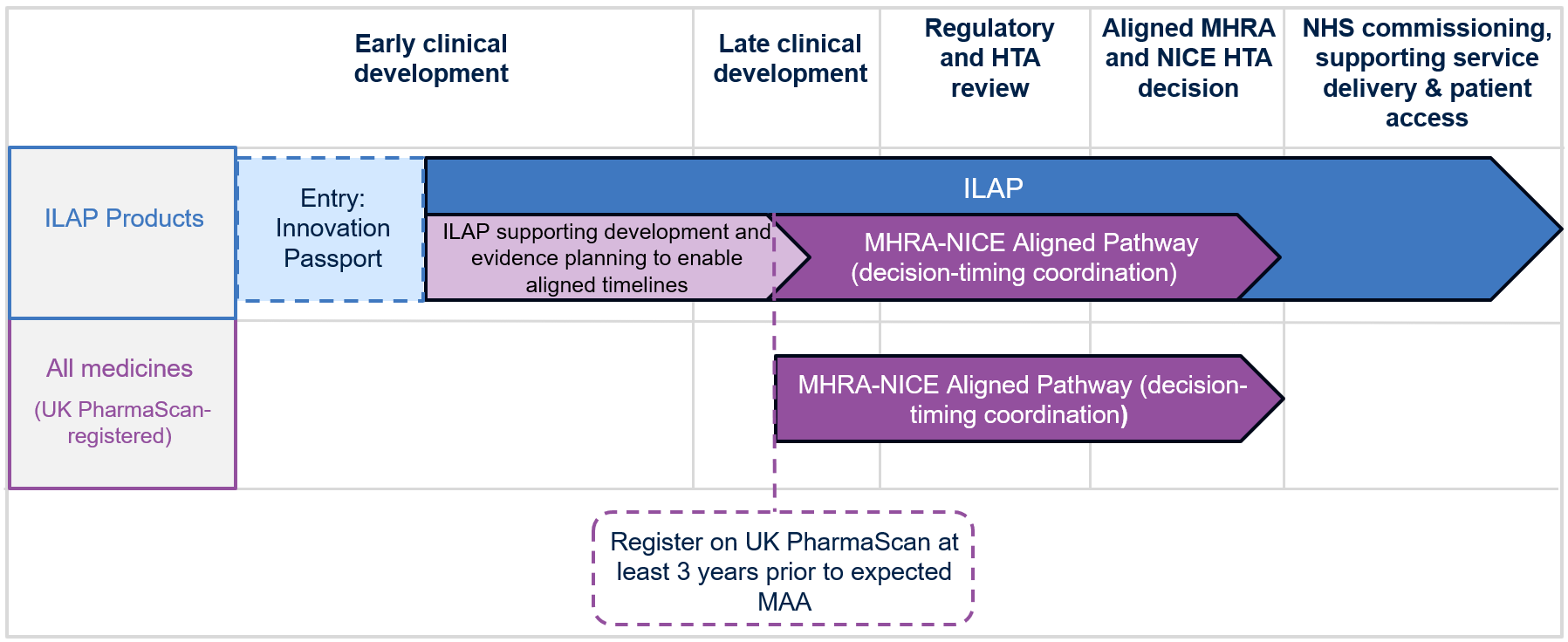
At the workshop, the MHRA expert, Ian Rees set out how the UK’s decentralised manufacturing framework is pushing boundaries on regulatory innovation by adapting to new environments – including in orbit while maintaining safety, quality and oversight. Watch Ian Rees discuss this work in a short video.
The workshop underscored the importance of international collaboration in building the evidence base for space-enabled life sciences. Participants highlighted the complementary strengths of the UK and Switzerland in scientific research, advanced manufacturing, and regulatory innovation, creating a strong foundation for future joint initiatives.
Discussions highlighted the importance of ongoing engagement between regulators, industry, academia and funding organisations to support the effective translation of emerging research into practice. Collaborative pilot programmes and knowledge-sharing partnerships were identified as valuable mechanisms for building evidence, reducing barriers to adoption and delivering meaningful healthcare benefits.
Clear regulatory pathways are essential for early pilots and future investment decisions. It also strengthens the UK’s position as a credible location for space-enabled life sciences activity.
"Innovation in space should be supported by regulation that is both robust and adaptable. Our experience with decentralised manufacturing demonstrates how established regulatory principles can evolve to accommodate new operating environments, helping innovators move forward responsibly." Ian Rees, MHRA
]]>“What made this workshop distinctive was bringing together senior leaders from biopharma, space, academia and government to explore an emerging field from both scientific and commercial perspectives. The discussion demonstrated the value of having regulators included from early on. The UK and Switzerland are well placed to collaborate through their strengths in life sciences, space, research and innovation. We look forward to building on the momentum through further engagement and our forthcoming white paper.” Helen Stubbs, Department for Business and Trade, British Embassy Berne