When updating product licence details, there are always areas in the variation submission processes (both National and Mutual Recognition (MR)) which can cause validation issues or the generation of Notification with Grounds (NWG) letters, ultimately resulting in a rejection or delay. These can be time consuming for both the MAH (Marketing Authorisation Holder) and our variations team and may impact on assessment timeframes (Type 1B/II).

Many situations in the submission process can affect validation and areas range - from missing SmPC fragments (31%) to choosing the correct change code for the submission (8%). Proof of Payment was a major reason for delays and accounted for 32% of all early rejections before the change to ‘pay-on-invoice’, but more about that change later!
If a submission passes validation but does not contain all the necessary data for a full assessment, an NWG letter (sometimes known as a “Request for Further Information” or RFI) will be generated and depending on the procedure, may add 30-60 days to the expected assessment timeframe. At the moment, NWG letters are generated on more than 75% of varation applications.
We continually review our processes and over the last couple of years we have introduced measures to reduce both the regulatory burden on industry and processing impact on MHRA. These include fee calculators, paperless communication with immediate access to letters and more recently, the ‘pay-on-invoice’ process introduced on 1st April 2017.
We have also been looking at how the Agency has worked closely with industry to address issues and our interaction with the regulatory community. One way has been to pay close attention to feedback we receive from delegates who attend our various events. From this we can create and tailor future events to meet industry expectations based upon experiences and demand.
An example of how we have done this was the November 2016 Licensing Division ‘Abridged Symposium – Getting it right first time’. This meeting focussed on helping applicants minimise deficiencies in new Marketing Authorisation Applications.
Feedback was very positive, especially for those presentations that promoted minimising major quality deficiencies (drug substance and drug product) and the regulatory fundamentals of bioequivalence and biowaivers. Agency experts highlighted the key themes which have emerged over the last 5 years together with the main “show stoppers” (the top five main deficiencies are shown in the pie chart.) and gave advice on how to avoid them. Other topics covered included legal basis (well-established use, hybrids, and informed consent), advantages of scientific advice and drug-device combinations.
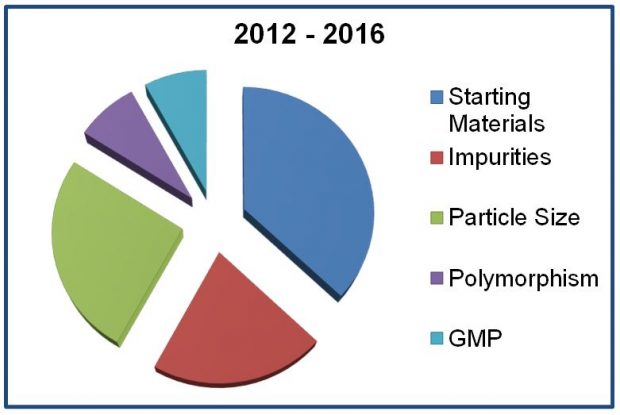
Feedback and delegate suggestions collected from this event highlighted areas where industry felt our expert assistance could help with planning submissions and, as the symposium title suggested, reduce the possibility of NWG/RFI letters.
The planning of events for the second half of 2017 is now underway. Over the coming months assessment teams will mull over quality and clinical subject matter to help further improve the submission and validation processes so that the data packages received as part of a submission, contain the correct information for a smooth assessment. This includes those areas around regulatory fundamentals leading to minimising deficiencies in new MAAs through to a workshop on variations with presentations on grouping and the Composite Coordinated Collection (CCC).
Articles lined up for posting in future blogs include ‘Hints & Tips’, ‘Help with Type 1B Safety Variations’ and more details about future events planned by the agency.
Sign up for new post alerts and let us know if there is anything else you'd like to know about any of our upcoming events.
2 comments
Comment by Beena Shaikh posted on
If the agency could share the common/ frequent queries raised during variation assessment for Quality changes then it would really help MAH for getting it right for the first time.Thanks
Comment by Sarah-Rose Burke posted on
Hi Beena, thanks for your comment. I will pass that on to our team and consider your suggestion for future posts.